Tutorial for Real-World Data
This tutorial offers a step-by-step guide to replicating the real-world results shown in our paper. Please make sure you have downloaded the required datasets from our repository before executing the code. Some important parameters in our model are:
interpretable: Whether or not to use interpretable mode of SpaMV.max_epoch_stage1: the maximum number of epoches for the first stage of SpaMV.max_epoch_stage2: the maximum number of epoches for the second stage of SpaMV (only useful for interpretable dimension reduction mode).weights: the hyperparameters that re-weight the reconstruction loss for different modalities. A higher value for one modality would lead the model values that modality more than others.alphas: the hyperparameters that regulate the strength of the uniqueness constraint on the private latent variables.
Domain Clustering on Real-World Data
In this section, we demonstrate the training procedure and clustering outcomes for the four real-world datasets featured in our paper. We applied distinct preprocessing methods for various omics data types, implemented in SpaMV.utils.preprocess_dc. If you wish to use your own preprocessing approach, simply substitute that line with your preferred method.
[1]:
from sklearn.metrics import adjusted_rand_score
from SpaMV.spamv import SpaMV
from SpaMV.utils import preprocess_dc, clustering
import scanpy as sc
import matplotlib.pyplot as plt
dataname = 'Mouse_Embryo'
datafolder = 'Data/' + dataname
scale=False
if dataname == 'Mouse_Embryo':
datasets = [sc.read_h5ad(datafolder + '/adata_RNA.h5ad'), sc.read_h5ad(datafolder + '/adata_peaks.h5ad')]
omics_names = ['Transcriptome', 'Epigenome']
n_cluster = 14
elif dataname == 'ME13_1':
datasets = [sc.read_h5ad(datafolder + '/adata_H3K27ac_ATAC.h5ad'),
sc.read_h5ad(datafolder + '/adata_H3K27me3_ATAC.h5ad')]
omics_names = ['H3K27ac', 'H3K27me3']
n_cluster = 21
elif dataname == 'Mouse_Thymus':
datasets = [sc.read_h5ad(datafolder + '/adata_RNA.h5ad'), sc.read_h5ad(datafolder + '/adata_ADT.h5ad')]
omics_names = ['Transcriptome', 'Proteome']
n_cluster = 7
scale=True
elif dataname == 'ccRCC_Y7_T':
datasets = [sc.read_h5ad(datafolder + '/adata_RNA.h5ad'), sc.read_h5ad(datafolder + '/adata_MET.h5ad')]
omics_names = ['Transcriptome', 'Metabolome']
n_cluster = 16
else:
raise NotImplementedError
datasets = preprocess_dc(datasets, omics_names, scale=scale)
model = SpaMV(datasets, interpretable=False)
model.train()
datasets[0].obsm['SpaMV'] = model.get_embedding()
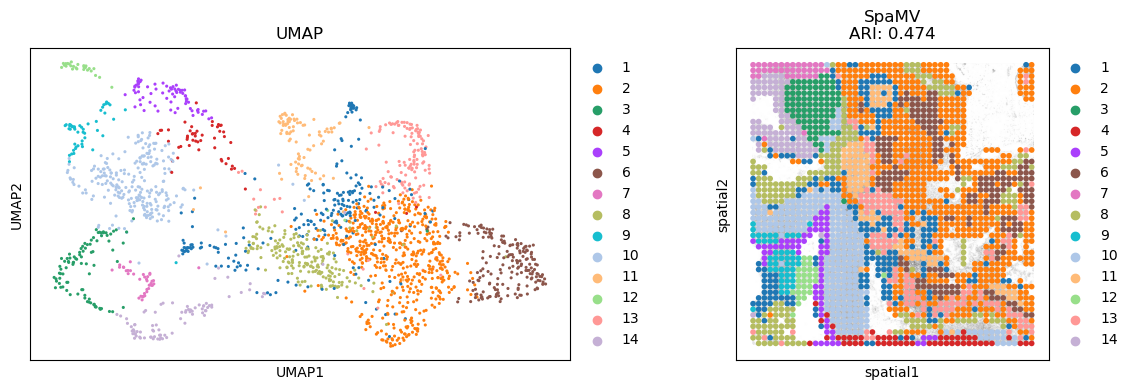
clustering(datasets[0], n_clusters=n_cluster, key='SpaMV')
fig, axes = plt.subplots(1, 2, figsize=(12, 4)) # 1 row, 2 columns
sc.pp.neighbors(datasets[0], use_rep='SpaMV')
sc.tl.umap(datasets[0])
sc.pl.umap(datasets[0], color='SpaMV', ax=axes[0], show=False, s=20, title='UMAP')
if 'spatial' in datasets[0].uns:
sc.pl.spatial(datasets[0], color='SpaMV', title='SpaMV\nARI: {:.3f}'.format(
adjusted_rand_score(datasets[0].obs['cluster'], datasets[0].obs['SpaMV'])) if 'cluster' in datasets[
0].obs else 'SpaMV', show=False, ax=axes[1])
else:
sc.pl.embedding(datasets[0], basis='spatial', color='SpaMV', show=False, ax=axes[1], size=50)
plt.tight_layout()
plt.show()
/home/comp/yangliu1214/anaconda3/envs/test/lib/python3.12/site-packages/dask/dataframe/__init__.py:31: FutureWarning: The legacy Dask DataFrame implementation is deprecated and will be removed in a future version. Set the configuration option `dataframe.query-planning` to `True` or None to enable the new Dask Dataframe implementation and silence this warning.
warnings.warn(
/home/comp/yangliu1214/anaconda3/envs/test/lib/python3.12/site-packages/xarray_schema/__init__.py:1: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import DistributionNotFound, get_distribution
/home/comp/yangliu1214/anaconda3/envs/test/lib/python3.12/site-packages/anndata/__init__.py:44: FutureWarning: Importing read_text from `anndata` is deprecated. Import anndata.io.read_text instead.
return module_get_attr_redirect(attr_name, deprecated_mapping=_DEPRECATED)
Building data and neighboring graphs...
100%|██████████| 2064/2064 [00:00<00:00, 65912.23it/s]
100%|██████████| 2064/2064 [00:00<00:00, 62297.92it/s]
Epoch Loss:315.889: 100%|██████████| 400/400 [01:04<00:00, 6.24it/s]
R callback write-console: __ __
____ ___ _____/ /_ _______/ /_
/ __ `__ \/ ___/ / / / / ___/ __/
/ / / / / / /__/ / /_/ (__ ) /_
/_/ /_/ /_/\___/_/\__,_/____/\__/ version 6.1.1
Type 'citation("mclust")' for citing this R package in publications.
fitting ...
|======================================================================| 100%
Interpretable Dimension Reduction on Real-World Data
[1]:
from matplotlib import pyplot as plt
from SpaMV.spamv import SpaMV
from SpaMV.utils import plot_embedding_results, preprocess_idr
import scanpy as sc
dataname = 'Mouse_Embryo'
data_folder = 'Data/' + dataname
zs_dim = 10
threshold_background = 1
weights = [1, 1]
alphas = [1, 1]
if dataname in ['Mouse_Thymus']:
data = [sc.read_h5ad(data_folder + '/adata_RNA.h5ad'), sc.read_h5ad(data_folder + '/adata_ADT.h5ad')]
omics_names = ['Transcriptome', 'Proteome']
zp_dims = [10, 10]
weights = [1, 10]
alphas = [5, 5]
elif dataname in ['Mouse_Embryo']:
data = [sc.read_h5ad(data_folder + '/adata_RNA.h5ad'), sc.read_h5ad(data_folder + '/adata_ATAC.h5ad')]
omics_names = ['Transcriptome', 'Epigenome']
zp_dims = [20, 5]
elif dataname in ['ME13_1']:
data = [sc.read_h5ad(data_folder + '/adata_H3K27ac_ATAC.h5ad'),
sc.read_h5ad(data_folder + '/adata_H3K27me3_ATAC.h5ad')]
omics_names = ['H3K27ac', 'H3K27me3']
zs_dim = 20
zp_dims = [5, 5]
alphas = [3, 3]
threshold_noise = .5
threshold_background = 5
elif dataname in ['ccRCC_Y7_T']:
data = [sc.read_h5ad(data_folder + '/adata_RNA.h5ad'), sc.read_h5ad(data_folder + '/adata_MET.h5ad')]
omics_names = ['Transcriptome', 'Metabolome']
zp_dims = [5, 5]
else:
raise Exception(f"Unkonwn Dataset: {dataname}")
data = preprocess_idr(data, omics_names)
model = SpaMV(data, zs_dim=zs_dim, zp_dims=zp_dims, interpretable=True, weights=weights, alphas=alphas,
omics_names=omics_names, threshold_background=threshold_background)
model.train()
z, w = model.get_embedding_and_feature_by_topic()
if dataname == 'ccRCC_Y7_T':
spatial_range = data[0].obsm['spatial'].min(0).tolist() + data[0].obsm['spatial'].max(0).tolist()
width = spatial_range[2] - spatial_range[0]
height = spatial_range[3] - spatial_range[1]
crop_coord = [(spatial_range[0] - width / 20, spatial_range[1] - height / 20, spatial_range[2] + width / 20,
spatial_range[3] + height / 20)]
plot_embedding_results(data, omics_names, z, w, show=True, save=False, crop_coord=crop_coord)
else:
plot_embedding_results(data, omics_names, z, w, show=True, save=False, size=100)
plt.show()
/home/comp/yangliu1214/anaconda3/envs/test/lib/python3.12/site-packages/dask/dataframe/__init__.py:31: FutureWarning: The legacy Dask DataFrame implementation is deprecated and will be removed in a future version. Set the configuration option `dataframe.query-planning` to `True` or None to enable the new Dask Dataframe implementation and silence this warning.
warnings.warn(
/home/comp/yangliu1214/anaconda3/envs/test/lib/python3.12/site-packages/xarray_schema/__init__.py:1: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import DistributionNotFound, get_distribution
/home/comp/yangliu1214/anaconda3/envs/test/lib/python3.12/site-packages/anndata/__init__.py:44: FutureWarning: Importing read_text from `anndata` is deprecated. Import anndata.io.read_text instead.
return module_get_attr_redirect(attr_name, deprecated_mapping=_DEPRECATED)
Building data and neighboring graphs...
100%|██████████| 2117/2117 [00:00<00:00, 6205.17it/s]
100%|██████████| 2117/2117 [00:00<00:00, 65852.90it/s]
Epoch Loss:1893.304: 100%|██████████| 400/400 [00:51<00:00, 7.81it/s]
Epoch Loss:1871.237: 100%|██████████| 400/400 [00:43<00:00, 9.17it/s]
merge Transcriptome private topic 4 and Transcriptome private topic 7

[ ]: